壓縮成 QIIME2 artifacts (.qza) 分析專用格式
在整個分析流程中,輸入的檔案必須轉換為 QIIME2 artifacts (.qza) 壓縮格式,這類型檔案之後會在一個一個分析流程中穿梭,而 .qza 是一種電腦看得懂,人類看不懂的玩意兒,a 指的就是人為加工過的檔案 (artifacts)。
可以想成將定序資料與註釋包裝的集合檔案。
那資料視覺化呢? 部分的 .qza 檔案有提供轉換為 .qzv功能,v 指的就是視覺化 (visualization) ,各種美美的圖都會從.qzv 跑出來! 變成電腦看不懂,人類看了很喜歡的玩意兒,因此接下來 .qza .qzv 兩個靈魂檔案格式會充斥本系列文 !
首先,必須先拎著 [Day 06] 所得到的三個檔案,
- 定序的原始檔案 (.fastq.gz)
- 樣本清單 manifest.tsv
- 註釋資料 sample-metadata.tsv
將三者轉換為一個 QIIME2 artifacts (.qza),幫助匯入,先啟動 qiime2-2022.8 環境 :
conda activate qiime2-2022.8匯入 (Importing)
第一步先將上述三個檔案匯入轉換為 .qza:
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-format PairedEndFastqManifestPhred33V2 \
--input-path manifest.tsv \
--output-path demux.qza import: 使用 qiime 軟體 插件為 tools 的 import 函式
–type: 16S 定序主流以 PairedEnd 為主 [詳情]
–input-format: Phred33V2 是一種對序列品質分數表示格式
–input-path: manifest.tsv 的所在路徑
–output-path: 轉換產出的 .qza 的檔名與路徑
實務上,定序檔案若是來自文獻、廠商,回到手上時多已拆分 (demultiplex) 完畢,所以跳過拆分步驟 (Barcode 部分我習慣在 QC 部分處理),以 demux.qza 命名,如有需要可以看這裡。
完成後會顯示:
"Imported manifest.tsv as PairedEndFastqManifestPhred33V2 to demux.qza"demux.qzv 視覺化 (visualization)
接下來我們將迎來第一個視覺化資料:
剛剛得到 demux.qza 轉為 .qzv,
輸入 :
qiime demux summarize \
--i-data demux.qza \
--o-visualization demux.qzv完成後會顯示 :
"Saved Visualization to: demux.qzv"將 demux.qzv 下載後拉到 QIIME VIEW 網站的框框,
就會跑出漂漂的 data ~ QIIME VIEW 可以直接加進我的最愛,之後會超常用到 !
資料解讀
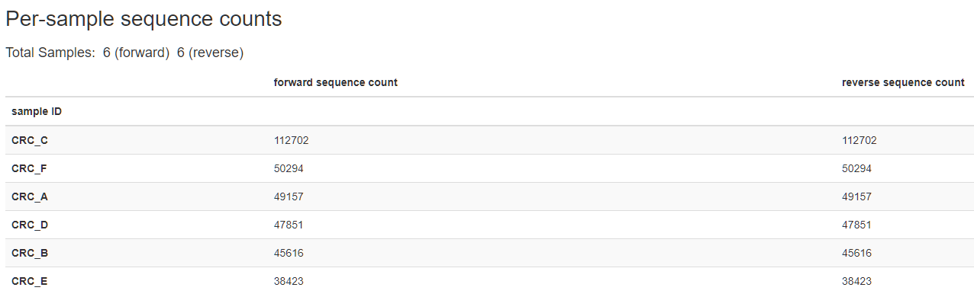
Overview 確認樣本名稱、數量與條數
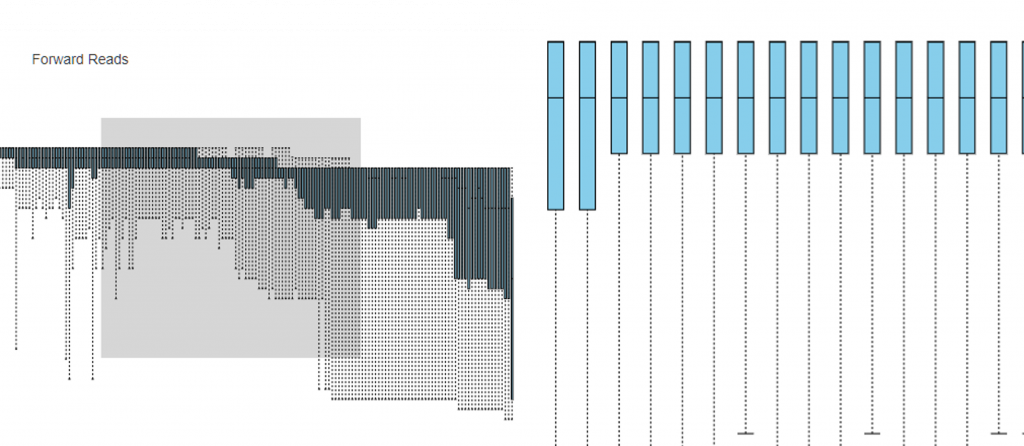
Interactive Quality Plot 序列品質查看
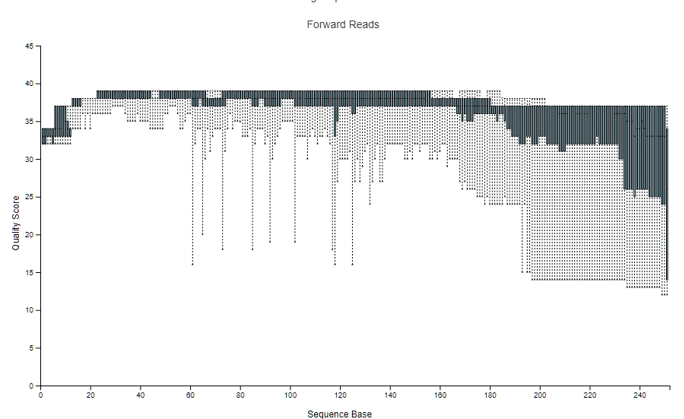
這個頁面非常重要,會影響到下一步要做的品質管制 (Quality Control),橫軸為每一條序列的長度,根據 第五篇文章 我們知道,序列長度取決於要 Primer 夾取哪個片段,範例中所使用的夾取 V3-V4 片段的 Primer (可以從 Paper 得知),長度大約是 470 bp。
在下一步前,先決定要切多少 : Trimming 修剪序列
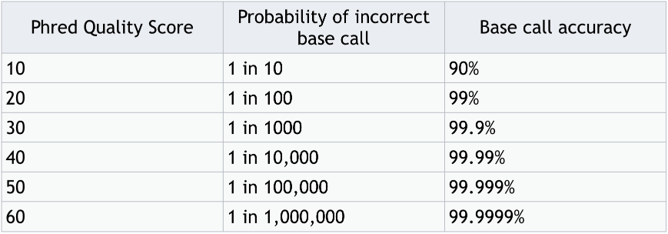
如果有些 base 的 Q Score < 25 怎麼辦呢 ?
這通常會發生在序列的一開始與結束,這時候就要決定要留多少的 nt,進行後續分析。 生科人如果曾經送過 Sanger 定序確認基因重組 / Primer夾取結果,像是加 His Tag, Plasmid 或是切膠定序確認:
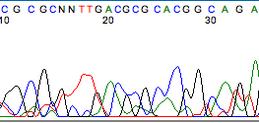
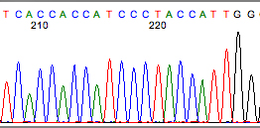
次世代定序也是如此,頭尾的訊號也會相較中段不穩定,因此需要進行修剪。
Reference
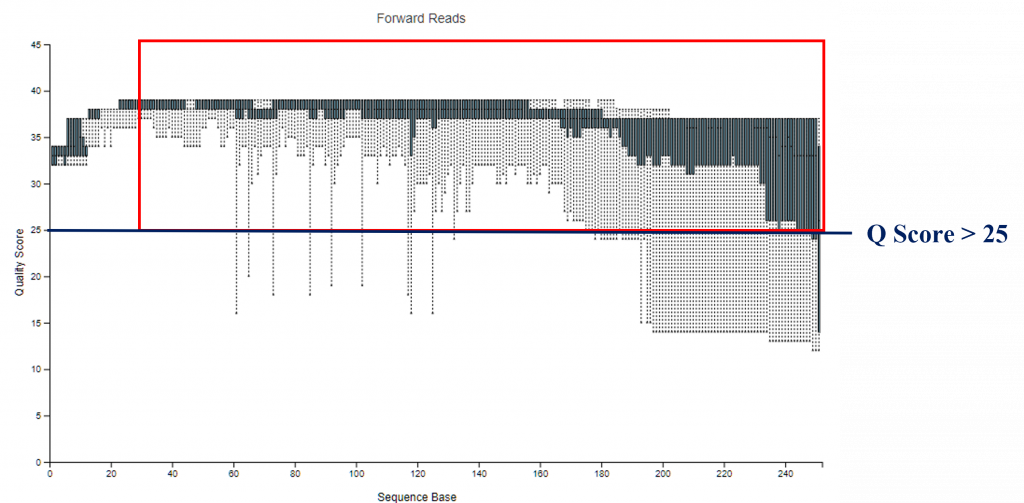
以範例來說,我習慣選擇 :
- 中位數 Q Score > 25 以上序列,
- 切除開頭前 10~30 nt,
實務上很常遇到不知道有無切除 Barcode,所以乾脆都切掉~
如果很肯定不含有,可以切 5~10 nt (Q Score < 25)即可。 - Forward、Reverse 盡量切等長,
避免後續 QC 兩序列無法互相組裝,
在這例子上,我們留下 10~250 nt ,抄在便條紙上,下回會用到。
本篇使用到的輸入/輸出檔案 :Input: .fastq.gz、manifest.tsv、sample-metadata.tsvOutput: demux.qza、demux.qzv
下回修剪序列與看品質管制 (Quality Control) 結果 !